Construct protein folding workflows to speed up drug discovery on Amazon SageMaker
Drug growth is a posh and lengthy course of that entails screening 1000’s of drug candidates and utilizing computational or experimental strategies to guage leads. According to McKinsey, a single drug can take 10 years and price a mean of $2.6 billion to undergo illness goal identification, drug screening, drug-target validation, and eventual industrial launch. Drug discovery is the analysis element of this pipeline that generates candidate medicine with the best chance of being efficient with the least hurt to sufferers. Machine studying (ML) strategies can assist establish appropriate compounds at every stage within the drug discovery course of, leading to extra streamlined drug prioritization and testing, saving billions in drug growth prices (for extra info, discuss with AI in biopharma research: A time to focus and scale).
Drug targets are sometimes organic entities referred to as proteins, the constructing blocks of life. The 3D construction of a protein determines the way it interacts with a drug compound; subsequently, understanding the protein 3D construction can add important enhancements to the drug growth course of by screening for drug compounds that match the goal protein construction higher. One other space the place protein construction prediction might be helpful is knowing the range of proteins, in order that we solely choose for medicine that selectively goal particular proteins with out affecting different proteins within the physique (for extra info, discuss with Improving target assessment in biomedical research: the GOT-IT recommendations). Exact 3D buildings of goal proteins can allow drug design with greater specificity and decrease chance of cross-interactions with different proteins.
Nonetheless, predicting how proteins fold into their 3D construction is a troublesome downside, and conventional experimental strategies akin to X-ray crystallography and NMR spectroscopy might be time-consuming and costly. Latest advances in deep studying strategies for protein analysis have proven promise in utilizing neural networks to foretell protein folding with outstanding accuracy. Folding algorithms like AlphaFold2, ESMFold, OpenFold, and RoseTTAFold can be utilized to shortly construct correct fashions of protein buildings. Sadly, these fashions are computationally costly to run and the outcomes might be cumbersome to match on the scale of 1000’s of candidate protein buildings. A scalable answer for utilizing these varied instruments will enable researchers and industrial R&D groups to shortly incorporate the most recent advances in protein construction prediction, handle their experimentation processes, and collaborate with analysis companions.
Amazon SageMaker is a completely managed service to arrange, construct, practice, and deploy high-quality ML fashions shortly by bringing collectively a broad set of capabilities purpose-built for ML. It provides a completely managed atmosphere for ML, abstracting away the infrastructure, knowledge administration, and scalability necessities so you may concentrate on constructing, coaching, and testing your fashions.
On this put up, we current a completely managed ML answer with SageMaker that simplifies the operation of protein folding construction prediction workflows. We first talk about the answer on the excessive stage and its person expertise. Subsequent, we stroll you thru find out how to simply arrange compute-optimized workflows of AlphaFold2 and OpenFold with SageMaker. Lastly, we display how one can monitor and evaluate protein construction predictions as a part of a typical evaluation. The code for this answer is out there within the following GitHub repository.
Resolution overview
On this answer, scientists can interactively launch protein folding experiments, analyze the 3D construction, monitor the job progress, and monitor the experiments in Amazon SageMaker Studio.
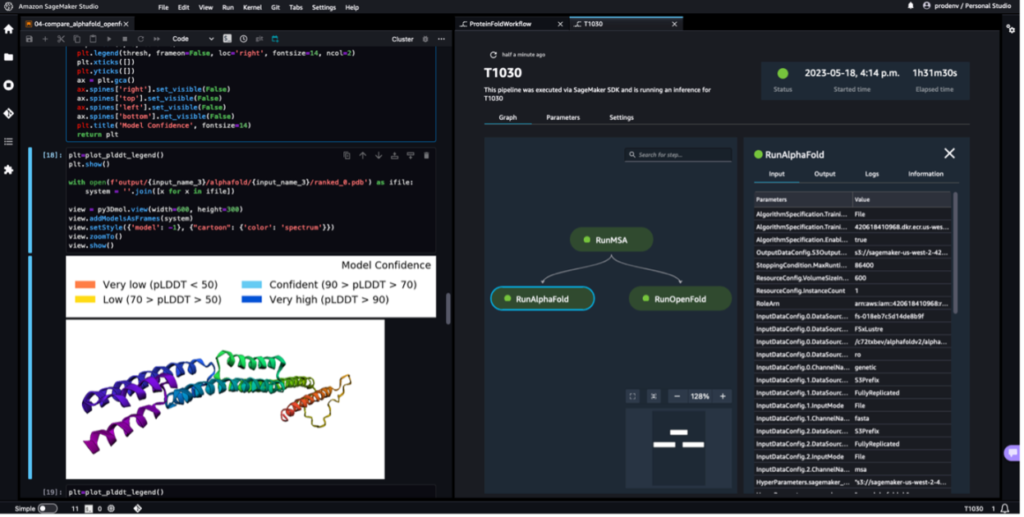
The next screenshot exhibits a single run of a protein folding workflow with Amazon SageMaker Studio. It contains the visualization of the 3D construction in a pocket book, run standing of the SageMaker jobs within the workflow, and hyperlinks to the enter parameters and output knowledge and logs.

The next diagram illustrates the high-level answer structure.

To grasp the structure, we first outline the important thing elements of a protein folding experiment as follows:
- FASTA goal sequence file – The FASTA format is a text-based format for representing both nucleotide sequences or amino acid (protein) sequences, during which nucleotides or amino acids are represented utilizing single-letter codes.
- Genetic databases – A genetic database is a number of units of genetic knowledge saved along with software program to allow customers to retrieve genetic knowledge. A number of genetic databases are required to run AlphaFold and OpenFold algorithms, akin to BFD, MGnify, PDB70, PDB, PDB seqres, UniRef30 (FKA UniClust30), UniProt, and UniRef90.
- A number of sequence alignment (MSA) – A sequence alignment is a manner of arranging the first sequences of a protein to establish areas of similarity which may be a consequence of practical, structural, or evolutionary relationships between the sequences. The enter options for predictions embrace MSA knowledge.
- Protein construction prediction – The construction of enter goal sequences is predicted with folding algorithms like AlphaFold2 and OpenFold that use a multitrack transformer structure skilled on recognized protein templates.
- Visualization and metrics – Visualize the 3D construction with the py3Dmol library as an interactive 3D visualization. You should use metrics to guage and evaluate construction predictions, most notably root-mean-square deviation (RMSD) and template modeling Score (TM-score)
The workflow comprises the next steps:
- Scientists use the web-based SageMaker ML IDE to discover the code base, construct protein sequence evaluation workflows in SageMaker Studio notebooks, and run protein folding pipelines by way of the graphical person interface in SageMaker Studio or the SageMaker SDK.
- Genetic and construction databases required by AlphaFold and OpenFold are downloaded previous to pipeline setup utilizing Amazon SageMaker Processing, an ephemeral compute function for ML knowledge processing, to an Amazon Simple Storage Service (Amazon S3) bucket. With SageMaker Processing, you may run a long-running job with a correct compute with out establishing any compute cluster and storage and without having to close down the cluster. Knowledge is robotically saved to a specified S3 bucket location.
- An Amazon FSx for Lustre file system is about up, with the information repository being the S3 bucket location the place the databases are saved. FSx for Lustre can scale to lots of of GB/s of throughput and thousands and thousands of IOPS with low-latency file retrieval. When beginning an estimator job, SageMaker mounts the FSx for Lustre file system to the occasion file system, then begins the script.
- Amazon SageMaker Pipelines is used to orchestrate a number of runs of protein folding algorithms. SageMaker Pipelines provides a desired visible interface for interactive job submission, traceability of the progress, and repeatability.
- Inside a pipeline, two computationally heavy protein folding algorithms—AlphaFold and OpenFold—are run with SageMaker estimators. This configuration helps mounting of an FSx for Lustre file system for top throughput database search within the algorithms. A single inference run is split into two steps: an MSA development step utilizing an optimum CPU occasion and a construction prediction step utilizing a GPU occasion. These substeps, like SageMaker Processing in Step 2, are ephemeral, on-demand, and absolutely managed. Job output akin to MSA recordsdata, predicted pdb construction recordsdata, and different metadata recordsdata are saved in a specified S3 location. A pipeline might be designed to run one single protein folding algorithm or run each AlphaFold and OpenFold after a typical MSA development.
- Runs of the protein folding prediction are robotically tracked by Amazon SageMaker Experiments for additional evaluation and comparability. The job logs are saved in Amazon CloudWatch for monitoring.
Conditions
To comply with this put up and run this answer, you’ll want to have accomplished a number of stipulations. Confer with the GitHub repository for an in depth clarification of every step.
Run protein folding on SageMaker
We use the absolutely managed capabilities of SageMaker to run computationally heavy protein folding jobs with out a lot infrastructure overhead. SageMaker makes use of container photos to run customized scripts for generic knowledge processing, coaching, and internet hosting. You may simply begin an ephemeral job on-demand that runs a program with a container picture with a few traces of the SageMaker SDK with out self-managing any compute infrastructure. Particularly, the SageMaker estimator job supplies flexibility in the case of selection of container picture, run script, and occasion configuration, and helps a wide variety of storage options, together with file methods akin to FSx for Lustre. The next diagram illustrates this structure.

Folding algorithms like AlphaFold and OpenFold use a multitrack transformer structure skilled on recognized protein templates to foretell the construction of unknown peptide sequences. These predictions might be run on GPU cases to supply greatest throughput and lowest latency. The enter options nevertheless for these predictions embrace MSA knowledge. MSA algorithms are CPU-dependent and may require a number of hours of processing time.
Operating each the MSA and construction prediction steps in the identical computing atmosphere might be cost-inefficient as a result of the costly GPU assets stay idle whereas the MSA step runs. Subsequently, we optimize the workflow into two steps. First, we run a SageMaker estimator job on a CPU occasion particularly to compute MSA alignment given a specific FASTA enter sequence and supply genetic databases. Then we run a SageMaker estimator job on a GPU occasion to foretell the protein construction with a given enter MSA alignment and a folding algorithm like AlphaFold or OpenFold.
Run MSA technology
For MSA computation, we embrace a customized script run_create_alignment.sh and create_alignments.py script that’s adopted from the prevailing AlphaFold prediction supply run_alphafold.py. Observe that this script could must be up to date if the supply AlphaFold code is up to date. The customized script is supplied to the SageMaker estimator by way of script mode. The important thing elements of the container picture, script mode implementation, and establishing a SageMaker estimator job are additionally a part of the following step of working folding algorithms, and are described additional within the following part.
Run AlphaFold
We get began by working an AlphaFold construction prediction with a single protein sequence utilizing SageMaker. Operating an AlphaFold job entails three easy steps, as might be seen in 01-run_stepbystep.ipynb. First, we construct a Docker container picture based mostly on AlphaFold’s Dockerfile in order that we will additionally run AlphaFold in SageMaker. Second, we assemble the script run_alphafold.sh that instructs how AlphaFold ought to be run. Third, we assemble and run a SageMaker estimator with the script, the container, occasion kind, knowledge, and configuration for the job.
Container picture
The runtime requirement for a container picture to run AlphaFold (OpenFold as effectively) in SageMaker might be significantly simplified with AlphaFold’s Dockerfile. We solely want so as to add a handful of easy layers on high to put in a SageMaker-specific Python library so {that a} SageMaker job can talk with the container picture. See the next code:
Enter script
We then present the script run_alphafold.sh that runs run_alphafold.py from the AlphaFold repository that’s at the moment positioned within the container /app/alphafold/run_alphafold.py. When this script is run, the situation of the genetic databases and the enter FASTA sequence can be populated by SageMaker as atmosphere variables (SM_CHANNEL_GENETIC and SM_CHANNEL_FASTA, respectively). For extra info, discuss with Input Data Configuration.
Estimator job
We subsequent create a job utilizing a SageMaker estimator with the next key enter arguments, which instruct SageMaker to run a selected script utilizing a specified container with the occasion kind or depend, your networking possibility of selection, and different parameters for the job. vpc_subnet_ids and security_group_ids instruct the job to run inside a selected VPC the place the FSx for Lustre file system is in in order that we will mount and entry the filesystem within the SageMaker job. The output path refers to a S3 bucket location the place the ultimate product of AlphaFold can be uploaded to on the finish of a profitable job by SageMaker robotically. Right here we additionally set a parameter DB_PRESET, for instance, to be handed in and accessed inside run_alphafold.sh as an environmental variable throughout runtime. See the next code:
from sagemaker.estimator import Estimator
alphafold_image_uri=f'{account}.dkr.ecr.{area}.amazonaws.com/sagemaker-studio-alphafold:v2.3.0'
instance_type="ml.g5.2xlarge"
instance_count=1
vpc_subnet_ids=['subnet-xxxxxxxxx'] # okay to make use of a default VPC
security_group_ids=['sg-xxxxxxxxx']
env={'DB_PRESET': db_preset} # <full_dbs|reduced_dbs>
output_path="s3://%s/%s/job-output/"%(default_bucket, prefix)
estimator_alphafold = Estimator(
source_dir="src", # listing the place run_alphafold.sh and different runtime recordsdata find
entry_point="run_alphafold.sh", # our script that runs /app/alphafold/run_alphafold.py
image_uri=alphafold_image_uri, # container picture to make use of
instance_count=instance_count, #
instance_type=instance_type,
subnets=vpc_subnet_ids,
security_group_ids=security_group_ids,
atmosphere=env,
output_path=output_path,
...)Lastly, we collect the information and let the job know the place they’re. The fasta knowledge channel is outlined as an S3 knowledge enter that can be downloaded from an S3 location into the compute occasion at the start of the job. This enables nice flexibility to handle and specify the enter sequence. Alternatively, the genetic knowledge channel is outlined as a FileSystemInput that can be mounted onto the occasion at the start of the job. The usage of an FSx for Lustre file system as a manner to herald shut to three TB of knowledge avoids repeatedly downloading knowledge from an S3 bucket to a compute occasion. We name the .match methodology to kick off an AlphaFold job:
from sagemaker.inputs import FileSystemInput
file_system_id='fs-xxxxxxxxx'
fsx_mount_id='xxxxxxxx'
file_system_directory_path=f'/{fsx_mount_id}/{prefix}/alphafold-genetic-db' # ought to be the total prefix from the S3 knowledge repository
file_system_access_mode="ro" # Specify the entry mode (read-only)
file_system_type="FSxLustre" # Specify your file system kind
genetic_db = FileSystemInput(
file_system_id=file_system_id,
file_system_type=file_system_type,
directory_path=file_system_directory_path,
file_system_access_mode=file_system_access_mode)
s3_fasta=sess.upload_data(path="sequence_input/T1030.fasta", # FASTA location domestically
key_prefix='alphafoldv2/sequence_input') # S3 prefix. Bucket is sagemaker default bucket
fasta = sagemaker.inputs.TrainingInput(s3_fasta,
distribution='FullyReplicated',
s3_data_type="S3Prefix",
input_mode="File")
data_channels_alphafold = {'genetic': genetic_db, 'fasta': fasta}
estimator_alphafold.match(inputs=data_channels_alphafold,
wait=False) # wait=False will get the cell again within the pocket book; set to True to see the logs because the job progressesThat’s it. We simply submitted a job to SageMaker to run AlphaFold. The logs and output together with .pdb prediction recordsdata can be written to Amazon S3.
Run OpenFold
Operating OpenFold in SageMaker follows the same sample, as proven within the second half of 01-run_stepbystep.ipynb. We first add a easy layer to get the SageMaker-specific library to make the container picture SageMaker appropriate on high of OpenFold’s Dockerfile. Secondly, we assemble a run_openfold.sh as an entry level for the SageMaker job. In run_openfold.sh, we run the run_pretrained_openfold.py from OpenFold, which is available in the container image with the identical genetic databases we downloaded for AlphaFold and OpenFold’s mannequin weights (--openfold_checkpoint_path). By way of enter knowledge places, in addition to the genetic databases channel and the FASTA channel, we introduce a 3rd channel, SM_CHANNEL_PARAM, in order that we will flexibly cross within the mannequin weights of selection from the estimator assemble after we outline and submit a job. With the SageMaker estimator, we will simply submit jobs with completely different entry_point, image_uri, atmosphere, inputs, and different configurations for OpenFold with the identical signature. For the information channel, we add a brand new channel, param, as an Amazon S3 enter together with using the identical genetic databases from the FSx for Lustre file system and FASTA file from Amazon S3. This, once more, permits us simply specify the mannequin weight to make use of from the job assemble. See the next code:
s3_param=sess.upload_data(path="openfold_params/finetuning_ptm_2.pt",
key_prefix=f'{prefix}/openfold_params')
param = sagemaker.inputs.TrainingInput(s3_param,
distribution="FullyReplicated",
s3_data_type="S3Prefix",
input_mode="File")
data_channels_openfold = {"genetic": genetic_db, 'fasta': fasta, 'param': param}
estimator_openfold.match(inputs=data_channels_openfold,
wait=False)To entry the ultimate output after the job completes, we run the next instructions:
!aws s3 cp {estimator_openfold.model_data} openfold_output/mannequin.tar.gz
!tar zxfv openfold_output/mannequin.tar.gz -C openfold_output/Runtime efficiency
The next desk exhibits the price financial savings of 57% and 51% for AlphaFold and OpenFold, respectively, by splitting the MSA alignment and folding algorithms in two jobs as in comparison with a single compute job. It permits us to right-size the compute for every job: ml.m5.4xlarge for MSA alignment and ml.g5.2xlarge for AlphaFold and OpenFold.
| Job Particulars | Occasion Sort | Enter FASTA Sequence | Runtime | Price |
| MSA alignment + OpenFold | ml.g5.4xlarge | T1030 | 50 minutes | $1.69 |
| MSA alignment + AlphaFold | ml.g5.4xlarge | T1030 | 65 minutes | $2.19 |
| MSA alignment | ml.m5.4xlarge | T1030 | 46 minutes | $0.71 |
| OpenFold | ml.g5.2xlarge | T1030 | 6 minutes | $0.15 |
| AlphaFold | ml.g5.2xlarge | T1030 | 21 minutes | $0.53 |
Construct a repeatable workflow utilizing SageMaker Pipelines
With SageMaker Pipelines, we will create an ML workflow that takes care of managing knowledge between steps, orchestrating their runs, and logging. SageMaker Pipelines additionally supplies us a UI to visualise our pipeline and simply run our ML workflow.
A pipeline is created by combing quite a lot of steps. On this pipeline, we mix three training steps, which require an SageMaker estimator. The estimators outlined on this pocket book are similar to these outlined in 01-run_stepbystep.ipynb, with the exception that we use Amazon S3 places to level to our inputs and outputs. The dynamic variables enable SageMaker Pipelines to run steps one after one other and likewise allow the person to retry failed steps. The next screenshot exhibits a Directed Acyclic Graph (DAG), which supplies info on the necessities for and relationships between every step of our pipeline.

Dynamic variables
SageMaker Pipelines is able to taking person inputs initially of each pipeline run. We outline the next dynamic variables, which we wish to change throughout every experiment:
- FastaInputS3URI – Amazon S3 URI of the FASTA file uploaded by way of SDK, Boto3, or manually.
- FastFileName – Identify of the FASTA file.
- db_preset – Choice between
full_dbsorreduced_dbs. - MaxTemplateDate – AlphaFold’s MSA step will seek for the obtainable templates earlier than the date specified by this parameter.
- ModelPreset – Choose between AlphaFold fashions together with
monomer,monomer_casp14,monomer_ptm, andmultimer. - NumMultimerPredictionsPerModel – Variety of seeds to run per mannequin when utilizing multimer system.
- InferenceInstanceType – Occasion kind to make use of for inference steps (each AlphaFold and OpenFold). The default worth is ml.g5.2xlarge.
- MSAInstanceType – Occasion kind to make use of for MSA step. The default worth is ml.m5.4xlarge.
See the next code:
fasta_file = ParameterString(identify="FastaFileName")
fasta_input = ParameterString(identify="FastaInputS3URI")
pipeline_db_preset = ParameterString(identify="db_preset",
default_value="full_dbs",
enum_values=['full_dbs', 'reduced_dbs'])
max_template_date = ParameterString(identify="MaxTemplateDate")
model_preset = ParameterString(identify="ModelPreset")
num_multimer_predictions_per_model = ParameterString(identify="NumMultimerPredictionsPerModel")
msa_instance_type = ParameterString(identify="MSAInstanceType", default_value="ml.m5.4xlarge")
instance_type = ParameterString(identify="InferenceInstanceType", default_value="ml.g5.2xlarge")A SageMaker pipeline is constructed by defining a sequence of steps after which chaining them collectively in a selected order the place the output of a earlier step turns into the enter to the following step. Steps might be run in parallel and outlined to have a dependency on a earlier step. On this pipeline, we outline an MSA step, which is the dependency for an AlphaFold inference step and OpenFold inference step that run in parallel. See the next code:
step_msa = TrainingStep(
identify="RunMSA",
step_args=pipeline_msa_args,
)
step_alphafold = TrainingStep(
identify="RunAlphaFold",
step_args=pipeline_alphafold_default_args,
)
step_alphafold.add_depends_on([step_msa])
step_openfold = TrainingStep(
identify="RunOpenFold",
step_args=pipeline_openfold_args,
)
step_openfold.add_depends_on([step_msa]To place all of the steps collectively, we name the Pipeline class and supply a pipeline identify, pipeline enter variables, and the person steps:
pipeline_name = f"ProteinFoldWorkflow"
pipeline = Pipeline(
identify=pipeline_name,
parameters=[
fasta_input,
instance_type,
msa_instance_type,
pipeline_db_preset
],
steps=[step_msa, step_alphafold, step_openfold],
)
pipeline.upsert(role_arn=function, # run this if it is the primary time establishing the pipeline
description='Protein_Workflow_MSA')Run the pipeline
Within the final cell of the pocket book 02-define_pipeline.ipynb, we present find out how to run a pipeline utilizing the SageMaker SDK. The dynamic variables we described earlier are supplied as follows:
!mkdir ./sequence_input/
!curl 'https://www.predictioncenter.org/casp14/goal.cgi?goal=T1030&view=sequence' > ./sequence_input/T1030.fasta
fasta_file_name="T1030.fasta"
pathName = f'./sequence_input/{fasta_file_name}'
s3_fasta=sess.upload_data(path=pathName,
key_prefix='alphafoldv2/sequence_input')
PipelineParameters={
'FastaInputS3URI':s3_fasta,
'db_preset': 'full_dbs',
'FastaFileName': fasta_file_name,
'MaxTemplateDate': '2020-05-14',
'ModelPreset': 'monomer',
'NumMultimerPredictionsPerModel': '5',
'InferenceInstanceType':'ml.g5.2xlarge',
'MSAInstanceType':'ml.m5.4xlarge'
}
execution = pipeline.begin(execution_display_name="SDK-Executetd",
execution_description='This pipeline was executed by way of SageMaker SDK',
parameters=PipelineParameters
)Monitor experiments and evaluate protein buildings
For our experiment, we use an instance protein sequence from the CASP14 competitors, which supplies an impartial mechanism for the evaluation of strategies of protein construction modeling. The goal T1030 is derived from the PDB 6P00 protein, and has 237 amino acids within the main sequence. We run the SageMaker pipeline to foretell the protein construction of this enter sequence with each OpenFold and AlphaFold algorithms.
When the pipeline is full, we obtain the anticipated .pdb recordsdata from every folding job and visualize the construction within the pocket book utilizing py3Dmol, as within the pocket book 04-compare_alphafold_openfold.ipynb.
The next screenshot exhibits the prediction from the AlphaFold prediction job.

The expected construction is in contrast towards its recognized base reference construction with PDB code 6poo archived in RCSB. We analyze the prediction efficiency towards the bottom PDB code 6poo with three metrics: RMSD, RMSD with superposition, and template modeling rating, as described in Comparing structures.
| . | Enter Sequence | Comparability With | RMSD | RMSD with Superposition | Template Modeling Rating |
| AlphaFold | T1030 | 6poo | 247.26 | 3.87 | 0.3515 |
The folding algorithms are actually in contrast towards one another for a number of FASTA sequences: T1030, T1090, and T1076. New goal sequences could not have the bottom pdb construction in reference databases and subsequently it’s helpful to match the variability between folding algorithms.
| . | Enter Sequence | Comparability With | RMSD | RMSD with Superposition | Template Modeling Rating |
| AlphaFold | T1030 | OpenFold | 73.21 | 24.8 | 0.0018 |
| AlphaFold | T1076 | OpenFold | 38.71 | 28.87 | 0.0047 |
| AlphaFold | T1090 | OpenFold | 30.03 | 20.45 | 0.005 |
The next screenshot exhibits the runs of ProteinFoldWorkflow for the three FASTA enter sequences with SageMaker Pipeline:

We additionally log the metrics with SageMaker Experiments as new runs of the identical experiment created by the pipeline:
from sagemaker.experiments.run import Run, load_run
metric_type="evaluate:"
experiment_name="proteinfoldworkflow"
with Run(experiment_name=experiment_name, run_name=input_name_1, sagemaker_session=sess) as run:
run.log_metric(identify=metric_type + "rmsd_cur", worth=rmsd_cur_one, step=1)
run.log_metric(identify=metric_type + "rmds_fit", worth=rmsd_fit_one, step=1)
run.log_metric(identify=metric_type + "tm_score", worth=tmscore_one, step=1)We then analyze and visualize these runs on the Experiments web page in SageMaker Studio.

The next chart depicts the RMSD worth between AlphaFold and OpenFold for the three sequences: T1030, T1076, and T1090.

Conclusion
On this put up, we described how you should use SageMaker Pipelines to arrange and run protein folding workflows with two widespread construction prediction algorithms: AlphaFold2 and OpenFold. We demonstrated a value performant answer structure of a number of jobs that separates the compute necessities for MSA technology from construction prediction. We additionally highlighted how one can visualize, consider, and evaluate predicted 3D buildings of proteins in SageMaker Studio.
To get began with protein folding workflows on SageMaker, discuss with the pattern code within the GitHub repo.
Concerning the authors
 Michael Hsieh is a Principal AI/ML Specialist Options Architect. He works with HCLS clients to advance their ML journey with AWS applied sciences and his experience in medical imaging. As a Seattle transplant, he loves exploring the good mom nature the town has to supply, such because the climbing trails, surroundings kayaking within the SLU, and the sundown at Shilshole Bay.
Michael Hsieh is a Principal AI/ML Specialist Options Architect. He works with HCLS clients to advance their ML journey with AWS applied sciences and his experience in medical imaging. As a Seattle transplant, he loves exploring the good mom nature the town has to supply, such because the climbing trails, surroundings kayaking within the SLU, and the sundown at Shilshole Bay.
 Shivam Patel is a Options Architect at AWS. He comes from a background in R&D and combines this along with his enterprise data to unravel advanced issues confronted by his clients. Shivam is most enthusiastic about workloads in machine studying, robotics, IoT, and high-performance computing.
Shivam Patel is a Options Architect at AWS. He comes from a background in R&D and combines this along with his enterprise data to unravel advanced issues confronted by his clients. Shivam is most enthusiastic about workloads in machine studying, robotics, IoT, and high-performance computing.
 Hasan Poonawala is a Senior AI/ML Specialist Options Architect at AWS, Hasan helps clients design and deploy machine studying functions in manufacturing on AWS. He has over 12 years of labor expertise as a knowledge scientist, machine studying practitioner, and software program developer. In his spare time, Hasan likes to discover nature and spend time with family and friends.
Hasan Poonawala is a Senior AI/ML Specialist Options Architect at AWS, Hasan helps clients design and deploy machine studying functions in manufacturing on AWS. He has over 12 years of labor expertise as a knowledge scientist, machine studying practitioner, and software program developer. In his spare time, Hasan likes to discover nature and spend time with family and friends.
 Jasleen Grewal is a Senior Utilized Scientist at Amazon Net Providers, the place she works with AWS clients to unravel actual world issues utilizing machine studying, with particular concentrate on precision medication and genomics. She has a powerful background in bioinformatics, oncology, and scientific genomics. She is enthusiastic about utilizing AI/ML and cloud providers to enhance affected person care.
Jasleen Grewal is a Senior Utilized Scientist at Amazon Net Providers, the place she works with AWS clients to unravel actual world issues utilizing machine studying, with particular concentrate on precision medication and genomics. She has a powerful background in bioinformatics, oncology, and scientific genomics. She is enthusiastic about utilizing AI/ML and cloud providers to enhance affected person care.





