Speed up protein construction prediction with the ESMFold language mannequin on Amazon SageMaker
Proteins drive many organic processes, comparable to enzyme exercise, molecular transport, and mobile help. The three-dimensional construction of a protein supplies perception into its perform and the way it interacts with different biomolecules. Experimental strategies to find out protein construction, comparable to X-ray crystallography and NMR spectroscopy, are costly and time-consuming.
In distinction, recently-developed computational strategies can quickly and precisely predict the construction of a protein from its amino acid sequence. These strategies are essential for proteins which can be troublesome to check experimentally, comparable to membrane proteins, the targets of many medicine. One well-known instance of that is AlphaFold, a deep learning-based algorithm celebrated for its correct predictions.
ESMFold is one other highly-accurate, deep learning-based methodology developed to foretell protein construction from its amino acid sequence. ESMFold makes use of a big protein language mannequin (pLM) as a spine and operates finish to finish. Not like AlphaFold2, it doesn’t want a lookup or Multiple Sequence Alignment (MSA) step, nor does it depend on exterior databases to generate predictions. As an alternative, the event workforce skilled the mannequin on thousands and thousands of protein sequences from UniRef. Throughout coaching, the mannequin developed consideration patterns that elegantly characterize the evolutionary interactions between amino acids within the sequence. This use of a pLM as a substitute of an MSA permits as much as 60 occasions quicker prediction occasions than different state-of-the-art fashions.
On this put up, we use the pre-trained ESMFold mannequin from Hugging Face with Amazon SageMaker to foretell the heavy chain construction of trastuzumab, a monoclonal antibody first developed by Genentech for the therapy of HER2-positive breast cancer. Shortly predicting the construction of this protein may very well be helpful if researchers wished to check the impact of sequence modifications. This might probably result in improved affected person survival or fewer unwanted side effects.
This put up supplies an instance Jupyter pocket book and associated scripts within the following GitHub repository.
Stipulations
We suggest operating this instance in an Amazon SageMaker Studio notebook operating the PyTorch 1.13 Python 3.9 CPU-optimized picture on an ml.r5.xlarge occasion kind.
Visualize the experimental construction of trastuzumab
To start, we use the biopython library and a helper script to obtain the trastuzumab construction from the RCSB Protein Data Bank:
Subsequent, we use the py3Dmol library to visualise the construction as an interactive 3D visualization:
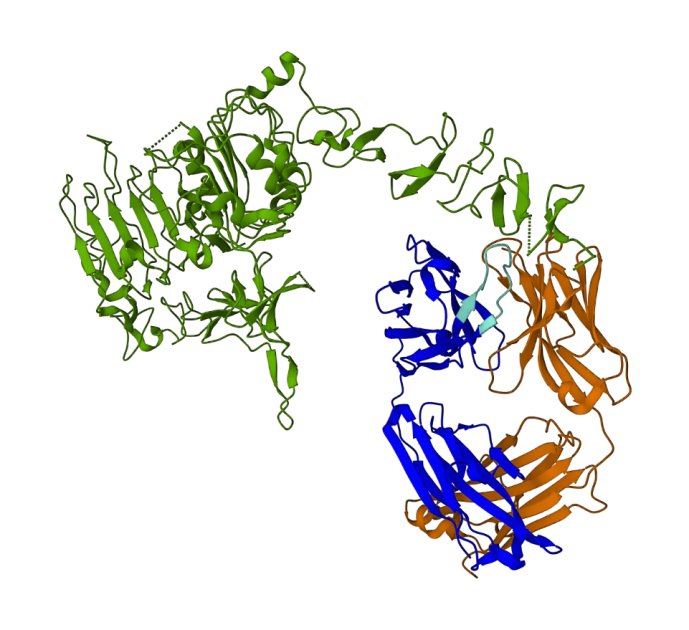
The next determine represents the 3D protein construction 1N8Z from the Protein Knowledge Financial institution (PDB). On this picture, the trastuzumab mild chain is displayed in orange, the heavy chain is blue (with the variable area in mild blue), and the HER2 antigen is inexperienced.
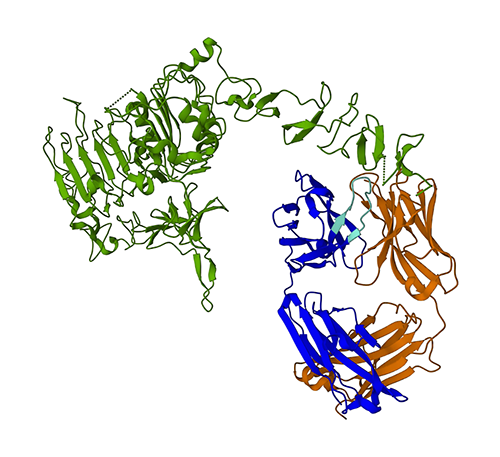
We’ll first use ESMFold to foretell the construction of the heavy chain (Chain B) from its amino acid sequence. Then, we are going to evaluate the prediction to the experimentally decided construction proven above.
Predict the trastuzumab heavy chain construction from its sequence utilizing ESMFold
Let’s use the ESMFold mannequin to foretell the construction of the heavy chain and evaluate it to the experimental end result. To begin, we’ll use a pre-built pocket book setting in Studio that comes with a number of essential libraries, like PyTorch, pre-installed. Though we may use an accelerated occasion kind to enhance the efficiency of our pocket book evaluation, we’ll as a substitute use a non-accelerated occasion and run the ESMFold prediction on a CPU.
First, we load the pre-trained ESMFold mannequin and tokenizer from Hugging Face Hub:
Subsequent, we copy the mannequin to our gadget (CPU on this case) and set some mannequin parameters:
To organize the protein sequence for evaluation, we have to tokenize it. This interprets the amino acid symbols (EVQLV…) right into a numerical format that the ESMFold mannequin can perceive (6,19,5,10,19,…):
Subsequent, we copy the tokenized enter to the mode, make a prediction, and save the end result to a file:
This takes about 3 minutes on a non-accelerated occasion kind, like a r5.
We are able to verify the accuracy of the ESMFold prediction by evaluating it to the experimental construction. We do that utilizing the US-Align device developed by the Zhang Lab on the College of Michigan:
| PDBchain1 | PDBchain2 | TM-Rating |
| information/prediction.pdb:A | information/experimental.pdb:B | 0.802 |
The template modeling score (TM-score) is a metric for assessing the similarity of protein constructions. A rating of 1.0 signifies an ideal match. Scores above 0.7 point out that proteins share the identical spine construction. Scores above 0.9 point out that the proteins are functionally interchangeable for downstream use. In our case of reaching TM-Rating 0.802, the ESMFold prediction would doubtless be acceptable for purposes like construction scoring or ligand binding experiments, however will not be appropriate to be used circumstances like molecular replacement that require extraordinarily excessive accuracy.
We are able to validate this end result by visualizing the aligned constructions. The 2 constructions present a excessive, however not good, diploma of overlap. Protein construction predictions is a rapidly-evolving area and plenty of analysis groups are creating ever-more correct algorithms!
Deploy ESMFold as a SageMaker inference endpoint
Operating mannequin inference in a pocket book is okay for experimentation, however what if you must combine your mannequin with an utility? Or an MLOps pipeline? On this case, a greater choice is to deploy your mannequin as an inference endpoint. Within the following instance, we’ll deploy ESMFold as a SageMaker real-time inference endpoint on an accelerated occasion. SageMaker real-time endpoints present a scalable, cost-effective, and safe strategy to deploy and host machine studying (ML) fashions. With automated scaling, you’ll be able to modify the variety of situations operating the endpoint to satisfy the calls for of your utility, optimizing prices and making certain excessive availability.
The pre-built SageMaker container for Hugging Face makes it simple to deploy deep studying fashions for widespread duties. Nonetheless, for novel use circumstances like protein construction prediction, we have to outline a customized inference.py script to load the mannequin, run the prediction, and format the output. This script consists of a lot of the identical code we utilized in our pocket book. We additionally create a necessities.txt file to outline some Python dependencies for our endpoint to make use of. You possibly can see the recordsdata we created within the GitHub repository.
Within the following determine, the experimental (blue) and predicted (purple) constructions of the trastuzumab heavy chain are very related, however not an identical.

After we’ve created the required recordsdata within the code listing, we deploy our mannequin utilizing the SageMaker HuggingFaceModel class. This makes use of a pre-built container to simplify the method of deploying Hugging Face fashions to SageMaker. Word that it could take 10 minutes or extra to create the endpoint, relying on the provision of ml.g4dn occasion varieties in our Area.
When the endpoint deployment is full, we are able to resubmit the protein sequence and show the primary few rows of the prediction:
As a result of we deployed our endpoint to an accelerated occasion, the prediction ought to solely take a number of seconds. Every row within the end result corresponds to a single atom and consists of the amino acid id, three spatial coordinates, and a pLDDT score representing the prediction confidence at that location.
| PDB_GROUP | ID | ATOM_LABEL | RES_ID | CHAIN_ID | SEQ_ID | CARTN_X | CARTN_Y | CARTN_Z | OCCUPANCY | PLDDT | ATOM_ID |
| ATOM | 1 | N | GLU | A | 1 | 14.578 | -19.953 | 1.47 | 1 | 0.83 | N |
| ATOM | 2 | CA | GLU | A | 1 | 13.166 | -19.595 | 1.577 | 1 | 0.84 | C |
| ATOM | 3 | CA | GLU | A | 1 | 12.737 | -18.693 | 0.423 | 1 | 0.86 | C |
| ATOM | 4 | CB | GLU | A | 1 | 12.886 | -18.906 | 2.915 | 1 | 0.8 | C |
| ATOM | 5 | O | GLU | A | 1 | 13.417 | -17.715 | 0.106 | 1 | 0.83 | O |
| ATOM | 6 | cg | GLU | A | 1 | 11.407 | -18.694 | 3.2 | 1 | 0.71 | C |
| ATOM | 7 | cd | GLU | A | 1 | 11.141 | -18.042 | 4.548 | 1 | 0.68 | C |
| ATOM | 8 | OE1 | GLU | A | 1 | 12.108 | -17.805 | 5.307 | 1 | 0.68 | O |
| ATOM | 9 | OE2 | GLU | A | 1 | 9.958 | -17.767 | 4.847 | 1 | 0.61 | O |
| ATOM | 10 | N | VAL | A | 2 | 11.678 | -19.063 | -0.258 | 1 | 0.87 | N |
| ATOM | 11 | CA | VAL | A | 2 | 11.207 | -18.309 | -1.415 | 1 | 0.87 | C |
Utilizing the identical methodology as earlier than, we see that the pocket book and endpoint predictions are an identical.
| PDBchain1 | PDBchain2 | TM-Rating |
| information/endpoint_prediction.pdb:A | information/prediction.pdb:A | 1.0 |
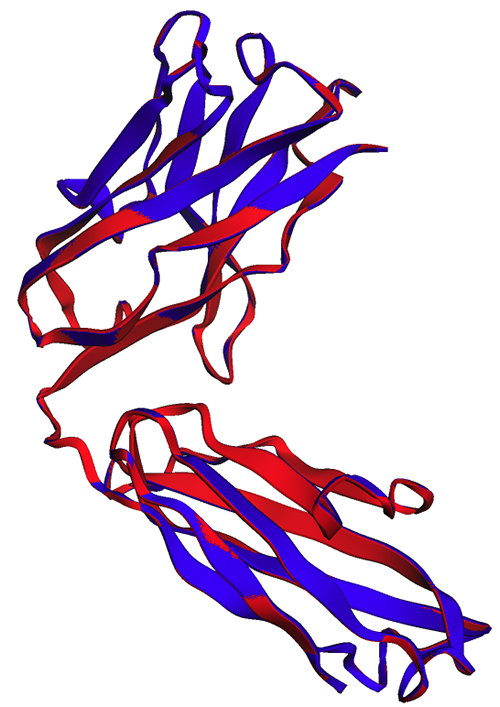
As noticed within the following determine, the ESMFold predictions generated in-notebook (purple) and by the endpoint (blue) present good alignment.
Clear up
To keep away from additional expenses, we delete our inference endpoint and take a look at information:
Abstract
Computational protein construction prediction is a essential device for understanding the perform of proteins. Along with fundamental analysis, algorithms like AlphaFold and ESMFold have many purposes in drugs and biotechnology. The structural insights generated by these fashions assist us higher perceive how biomolecules work together. This may then result in higher diagnostic instruments and therapies for sufferers.
On this put up, we present how one can deploy the ESMFold protein language mannequin from Hugging Face Hub as a scalable inference endpoint utilizing SageMaker. For extra details about deploying Hugging Face fashions on SageMaker, consult with Use Hugging Face with Amazon SageMaker. You may also discover extra protein science examples within the Awesome Protein Analysis on AWS GitHub repo. Please depart us a remark if there are every other examples you’d wish to see!
In regards to the Authors
 Brian Loyal is a Senior AI/ML Options Architect within the World Healthcare and Life Sciences workforce at Amazon Net Providers. He has greater than 17 years’ expertise in biotechnology and machine studying, and is keen about serving to prospects resolve genomic and proteomic challenges. In his spare time, he enjoys cooking and consuming along with his family and friends.
Brian Loyal is a Senior AI/ML Options Architect within the World Healthcare and Life Sciences workforce at Amazon Net Providers. He has greater than 17 years’ expertise in biotechnology and machine studying, and is keen about serving to prospects resolve genomic and proteomic challenges. In his spare time, he enjoys cooking and consuming along with his family and friends.
 Shamika Ariyawansa is an AI/ML Specialist Options Architect within the World Healthcare and Life Sciences workforce at Amazon Net Providers. He passionately works with prospects to speed up their AI and ML adoption by offering technical steering and serving to them innovate and construct safe cloud options on AWS. Exterior of labor, he loves snowboarding and off-roading.
Shamika Ariyawansa is an AI/ML Specialist Options Architect within the World Healthcare and Life Sciences workforce at Amazon Net Providers. He passionately works with prospects to speed up their AI and ML adoption by offering technical steering and serving to them innovate and construct safe cloud options on AWS. Exterior of labor, he loves snowboarding and off-roading.
 Yanjun Qi is a Senior Utilized Science Supervisor on the AWS Machine Studying Answer Lab. She innovates and applies machine studying to assist AWS prospects pace up their AI and cloud adoption.
Yanjun Qi is a Senior Utilized Science Supervisor on the AWS Machine Studying Answer Lab. She innovates and applies machine studying to assist AWS prospects pace up their AI and cloud adoption.