CRISPR-Cas9 information RNA effectivity prediction with effectively tuned fashions in Amazon SageMaker
The clustered commonly interspaced brief palindromic repeat (CRISPR) know-how holds the promise to revolutionize gene modifying applied sciences, which is transformative to the best way we perceive and deal with illnesses. This method is predicated in a pure mechanism present in micro organism that permits a protein coupled to a single information RNA (gRNA) strand to find and make cuts in particular websites within the focused genome. Having the ability to computationally predict the effectivity and specificity of gRNA is central to the success of gene modifying.
Transcribed from DNA sequences, RNA is a vital kind of organic sequence of ribonucleotides (A, U, G, C), which folds into 3D construction. Benefiting from latest advance in giant language fashions (LLMs), quite a lot of computational biology duties may be solved by fine-tuning organic LLMs pre-trained on billions of recognized organic sequences. The downstream duties on RNAs are comparatively understudied.
On this put up, we undertake a pre-trained genomic LLMs for gRNA effectivity prediction. The thought is to deal with a pc designed gRNA as a sentence, and fine-tune the LLM to carry out sentence-level regression duties analogous to sentiment evaluation. We used Parameter-Environment friendly Superb-Tuning strategies to cut back the variety of parameters and GPU utilization for this activity.
Answer overview
Massive language fashions (LLMs) have gained a variety of curiosity for his or her skill to encode syntax and semantics of pure languages. The neural structure behind LLMs are transformers, that are comprised of attention-based encoder-decoder blocks that generate an inner illustration of the info they’re skilled from (encoder) and are in a position to generate sequences in the identical latent area that resemble the unique knowledge (decoder). Because of their success in pure language, latest works have explored the usage of LLMs for molecular biology data, which is sequential in nature.
DNABERT is a pre-trained transformer mannequin with non-overlapping human DNA sequence knowledge. The spine is a BERT structure made up of 12 encoding layers. The authors of this mannequin report that DNABERT is ready to seize an excellent characteristic illustration of the human genome that allows state-of-the-art efficiency on downstream duties like promoter prediction and splice/binding web site identification. We determined to make use of this mannequin as the inspiration for our experiments.
Regardless of the success and common adoption of LLMs, fine-tuning these fashions may be troublesome due to the variety of parameters and computation essential for it. Because of this, Parameter-Environment friendly Superb-Tuning (PEFT) strategies have been developed. On this put up, we use certainly one of these strategies, referred to as LoRA (Low-Rank Adaptation). We introduce the strategy within the following sections.
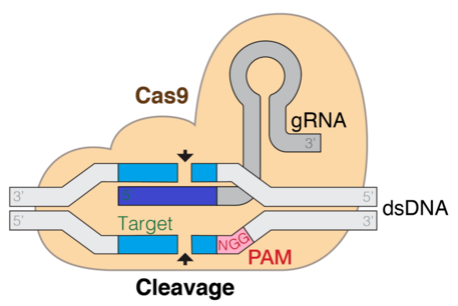
The next diagram is a illustration of the Cas9 DNA goal mechanism. The gRNA is the element that helps goal the cleavage web site.

The aim of this resolution is to fine-tune a base DNABERT mannequin to foretell exercise effectivity from totally different gRNA candidates. As such, our resolution first takes gRNA knowledge and processes it, as described later on this put up. Then we use an Amazon SageMaker notebook and the Hugging Face PEFT library to fine-tune the DNABERT mannequin with the processed RNA knowledge. The label we need to predict is the effectivity rating because it was calculated in experimental circumstances testing with the precise RNA sequences in cell cultures. These scores describe a stability between having the ability to edit the genome and never harm DNA that wasn’t focused.
The next diagram illustrates the workflow of the proposed resolution.

Conditions
For this resolution, you want entry to the next:
- A SageMaker pocket book occasion (we skilled the mannequin on an ml.g4dn.8xlarge occasion with a single NVIDIA T4 GPU)
- transformers-4.34.1
- peft-0.5.0
- DNABERT 6
Dataset
For this put up, we use the gRNA knowledge launched by researchers in a paper about gRNA prediction using deep learning. This dataset accommodates effectivity scores calculated for various gRNAs. On this part, we describe the method we adopted to create the coaching and analysis datasets for this activity.
To coach the mannequin, you want a 30-mer gRNA sequence and effectivity rating. A k-mer is a contiguous sequence of okay nucleotide bases extracted from an extended DNA or RNA sequence. For instance, when you’ve got the DNA sequence “ATCGATCG” and also you select okay = 3, then the k-mers inside this sequence can be “ATC,” “TCG,” “CGA,” “GAT,” and “ATC.”
Effectivity rating
Begin with excel file 41467_2021_23576_MOESM4_ESM.xlsx from the CRISPRon paper within the Supplementary Knowledge 1 part. On this file, the authors launched the gRNA (20-mer) sequences and corresponding total_indel_eff scores. We particularly used the info from the sheet named spCas9_eff_D10+dox. We use the total_indel_eff column because the effectivity rating.
Coaching and validation knowledge
Given the 20-mers and the crispron scores (similar because the total_indel_eff scores) from earlier, full the next steps to place collectively the coaching and validation knowledge:
- Convert the sequences within the sheet “TRAP12K microarray oligos” into an .fa (fasta) file.
- Run the script
get_30mers_from_fa.py(from the CRISPRon GitHub repository) to acquire all potential 23-mers and 30-mers from the sequences obtained from Step 1. - Use the
CRISPRspec_CRISPRoff_pipeline.pyscript (from the CRISPRon GitHub repository) to acquire the binding vitality for the 23-mers obtained from Step 2. For extra particulars on the way to run this script, try the code launched by the authors of the CRISPRon paper(verify the scriptCRISPRon.sh). - At this level, we now have 23-mers together with the corresponding binding vitality scores, and 20-mers together with the corresponding CRISPRon scores. Moreover, we now have the 30-mers from Step 2.
- Use the script
prepare_train_dev_data.py(from our launched code) to create coaching and validation splits. Operating this script will create two recordsdata:prepare.csvanddev.csv.
The information appears to be like one thing like the next:
Mannequin structure for gRNA encoding
To encode the gRNA sequence, we used the DNABERT encoder. DNABERT was pre-trained on human genomic knowledge, so it’s an excellent mannequin to encode gRNA sequences. DNABERT tokenizes the nucleotide sequence into overlapping k-mers, and every k-mer serves as a phrase within the DNABERT mannequin’s vocabulary. The gRNA sequence is damaged right into a sequence of k-mers, after which every k-mer is changed by an embedding for the k-mer on the enter layer. In any other case, the structure of DNABERT is much like that of BERT. After we encode the gRNA, we use the illustration of the [CLS] token as the ultimate encoding of the gRNA sequence. To foretell the effectivity rating, we use an extra regression layer. The MSE loss would be the coaching goal. The next is a code snippet of the DNABertForSequenceClassification mannequin:
Superb-tuning and prompting genomic LLMs
Superb-tuning all of the parameters of a mannequin is dear as a result of the pre-trained mannequin turns into a lot bigger. LoRA is an progressive approach developed to deal with the problem of fine-tuning extraordinarily giant language fashions. LoRA provides an answer by suggesting that the pre-trained mannequin’s weights stay fastened whereas introducing trainable layers (known as rank-decomposition matrices) inside every transformer block. This strategy considerably reduces the variety of parameters that have to be skilled and lowers the GPU reminiscence necessities, as a result of most mannequin weights don’t require gradient computations.
Subsequently, we adopted LoRA as a PEFT technique on the DNABERT mannequin. LoRA is applied within the Hugging Face PEFT library. When utilizing PEFT to coach a mannequin with LoRA, the hyperparameters of the low rank adaptation course of and the best way to wrap base transformers fashions may be outlined as follows:
Maintain-out analysis performances
We use RMSE, MSE, and MAE as analysis metrics, and we examined with rank 8 and 16. Moreover, we applied a easy fine-tuning technique, which is just including a number of dense layers after the DNABERT embeddings. The next desk summarizes the outcomes.
| Methodology | RMSE | MSE | MAE |
| LoRA (rank = 8) | 11.933 | 142.397 | 7.014 |
| LoRA (rank = 16) | 13.039 | 170.01 | 7.157 |
| One dense layer | 15.435 | 238.265 | 9.351 |
| Three dense layer | 15.435 | 238.241 | 9.505 |
| CRISPRon | 11.788 | 138.971 | 7.134 |
When rank=8, we now have 296,450 trainable parameters, which is about 33% trainable of the entire. The efficiency metrics are “rmse”: 11.933, “mse”: 142.397, “mae”: 7.014.
When rank=16, we now have 591,362 trainable parameters, which is about 66% trainable of the entire. The efficiency metrics are “rmse”: 13.039, “mse”: 170.010, “mae”: 7.157. There might need some overfitting challenge right here underneath this setting.
We additionally evaluate what occurs when including a number of dense layers:
- After including one dense layer, we now have “rmse”: 15.435, “mse”: 238.265, “mae”: 9.351
- After including three dense layers, we now have “rmse”: 15.435, “mse”: 238.241, “mae”: 9.505
Lastly, we evaluate with the present CRISPRon technique. CRISPRon is a CNN primarily based deep studying mannequin. The efficiency metrics are “rmse”: 11.788, “mse”: 138.971, “mae”: 7.134.
As anticipated, LoRA is doing significantly better than merely including a number of dense layers. Though the efficiency of LoRA is a bit worse than CRISPRon, with thorough hyperparameter search, it’s prone to outperform CRISPRon.
When utilizing SageMaker notebooks, you’ve the flexibleness to save lots of the work and knowledge produced in the course of the coaching, flip off the occasion, and switch it again on while you’re able to proceed the work, with out dropping any artifacts. Turning off the occasion will maintain you from incurring prices on compute you’re not utilizing. We extremely advocate solely turning it on while you’re actively utilizing it.
Conclusion
On this put up, we confirmed the way to use PEFT strategies for fine-tuning DNA language fashions utilizing SageMaker. We targeted on predicting effectivity of CRISPR-Cas9 RNA sequences for his or her affect in present gene-editing applied sciences. We additionally supplied code that may show you how to jumpstart your biology functions in AWS.
To study extra concerning the healthcare and life science area, confer with Run AlphaFold v2.0 on Amazon EC2 or fine-tuning Fine-tune and deploy the ProtBERT model for protein classification using Amazon SageMaker.
In regards to the Authors
 Siddharth Varia is an utilized scientist in AWS Bedrock. He’s broadly involved in pure language processing and has contributed to AWS merchandise similar to Amazon Comprehend. Exterior of labor, he enjoys exploring new locations and studying. He bought on this venture after studying the ebook The Code Breaker.
Siddharth Varia is an utilized scientist in AWS Bedrock. He’s broadly involved in pure language processing and has contributed to AWS merchandise similar to Amazon Comprehend. Exterior of labor, he enjoys exploring new locations and studying. He bought on this venture after studying the ebook The Code Breaker.
 Yudi Zhang is an Utilized Scientist at AWS advertising and marketing. Her analysis pursuits are within the space of graph neural networks, pure language processing, and statistics.
Yudi Zhang is an Utilized Scientist at AWS advertising and marketing. Her analysis pursuits are within the space of graph neural networks, pure language processing, and statistics.
 Erika Pelaez Coyotl is a Sr Utilized Scientist in Amazon Bedrock, the place she’s at the moment serving to develop the Amazon Titan giant language mannequin. Her background is in biomedical science, and he or she has helped a number of prospects develop ML fashions on this vertical.
Erika Pelaez Coyotl is a Sr Utilized Scientist in Amazon Bedrock, the place she’s at the moment serving to develop the Amazon Titan giant language mannequin. Her background is in biomedical science, and he or she has helped a number of prospects develop ML fashions on this vertical.
 Zichen Wang is a Sr Utilized Scientist in AWS AI Analysis & Schooling. He’s involved in researching graph neural networks and making use of AI to speed up scientific discovery, particularly on molecules and simulations.
Zichen Wang is a Sr Utilized Scientist in AWS AI Analysis & Schooling. He’s involved in researching graph neural networks and making use of AI to speed up scientific discovery, particularly on molecules and simulations.
 Rishita Anubhai is a Principal Utilized Scientist in Amazon Bedrock. She has deep experience in pure language processing and has contributed to AWS initiatives like Amazon Comprehend, Machine Studying Options Lab, and improvement of Amazon Titan fashions. She’s keenly involved in utilizing machine studying analysis, particularly deep studying, to create tangible affect.
Rishita Anubhai is a Principal Utilized Scientist in Amazon Bedrock. She has deep experience in pure language processing and has contributed to AWS initiatives like Amazon Comprehend, Machine Studying Options Lab, and improvement of Amazon Titan fashions. She’s keenly involved in utilizing machine studying analysis, particularly deep studying, to create tangible affect.